

The standardization of impurity studies and their control within a safe and reasonable limit will directly affect the quality and safety of marketed drugs. Let’s learn more about impurity studies with uHPLCs.
Source analysis of impurities
By introducing the concept of Quality by Design (QbD) into the study of drug impurities, the impurity profile of a product can be initially outlined before drug production based on the synthesis mechanism of the specific process, the starting materials, and the basic structure of each intermediate.
The analysis of the source of impurities is the basis for developing a drug impurity control strategy. In particular, when analyzing the source of toxic impurities, all reagents, intermediates, and by-products of the synthesis and production process should be analyzed to speculate on potential impurities that may arise and analyze the solid impurities present. After synthesizing the API, the active compound of the drug is analyzed for toxicity and no longer contains the “alerting structure.” Still, the genotoxicity of compounds containing the alerting structure needs to be considered when used in the manufacturing process.
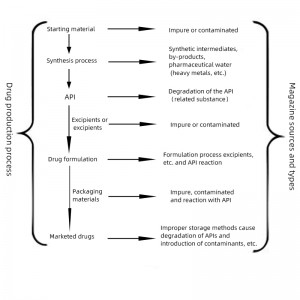
1. Classification of impurities and the existence of problems
Any substance affecting a drug’s purity is collectively referred to as an impurity. The study of impurities in drug development is carried out throughout its stages and is an important element in ensuring the quality of the drug. There are usually four ways to classify impurities in pharmaceuticals.
① Classification according to physical and chemical properties: organic impurities, inorganic impurities, and residual solvents.
② according to the source classification: process impurities ( including synthesis of incomplete reaction of reactants and reagents, intermediates, by-products, etc.), degradation products, impurities mixed in from the reactants and reagents, etc.
③ Classification by toxicity: toxic impurities and common impurities, etc.
④Classified according to chemical structure: other steroids, other alkaloids, geometric isomers, optical and polymers, etc. The presence of impurities greatly impacts the safety and efficacy of drugs, and how to set the limits of impurities is also a very sensitive issue in the drug development process. The main problem with impurity studies in China is that “the basis for setting impurity limits is insufficient.” They are usually determined by the research unit based on the results of multiple sample batches or so-called “experience.” In particular, the methodological studies are not comprehensive, the impurities are not effectively analyzed, and qualitative studies are conducted, resulting in imperfect impurity studies and large differences in quality between finished and foreign products.
2. General requirements
The development of pharmaceutical products needs to follow three principles: safety, effectiveness, and controlled quality. Therefore, when developing the limits of impurities in quality standards, the first consideration should be safety and effectiveness, especially for impurities with pharmacological activity or toxicity; the limits should be strictly controlled. Secondly, the feasibility of production and the normal fluctuations between batches should be considered, as well as the stability of the drug itself. Reasonable limits should be set according to the fluctuations in the process while ensuring safety and efficacy. In addition, the focus on different drugs or the same drug at different stages of research in the drug development process is different. Consequently, their impurity studies are unique and must be treated individually according to these specific circumstances.
Typically, impurity limits can be determined by establishing reporting, identification, and definition thresholds for degradation products based on the dose specified for the drug. Alternatively, impurity limits can be determined from toxicity data for known impurities or the results of toxicity studies.
(1) General requirements for ICH After the dose of the drug has been determined, the impurity limits can be established based on the threshold values for degradation products. In this case, impurity limits can be established by reference to the relevant requirements of the ICH (International Conference on Harmonisation of Technical Requirements for Registration of Pharmaceutical Products for Human Use).
For example, according to ICH requirements, for new APIs, the threshold for defining individual impurities is 0.05% for maximum daily doses greater than 2 g; for maximum daily doses not greater than 2 g, the threshold for defining impurities is 0.1% or 1 mg (according to strict requirements). When the maximum daily dose of an API is 1.5 g, the total intake of impurities at a defined threshold of 0.1% is 1.5 g x 0.1% = 1.5 mg, which is greater than the defined threshold of 1 mg.
If the threshold value is 1 mg, the limit for impurities is 1 mg ÷ 1.5 g x 100% = 0.07%, which is inconsistent with the defined threshold value (0.1%). In this case, the principle for setting impurity limits is “as strict as required,” i.e., the impurity limit should be set at 0.07%.
Of course, ICH is only a general requirement. Still, in the case of specific impurities (with a certain level of activity) or impurity limits exceeding the ICH requirement, relevant safety studies or literature data should be conducted to determine the reasonableness of the limit.
(2) Toxicity data for known impurity compounds
For known impurity compounds, toxicity data can be obtained from the relevant literature or several databases (e.g., www.discoveryGate.com), and the safety limits of the drug can be extrapolated from the proposed clinical dose of the drug.
For example, the main clinical toxic effects of impurity phenylhydrazine are acute intravascular hemorrhage, multiple damages to internal organs including the liver, kidney, heart, bone marrow, and central nervous system, and, at high concentrations, damage to lung tissue; even as a phenylhydrazine derivative, there is a potential risk of carcinogenicity. The literature[3] shows that the administration of phenylhydrazine at 0.6, 6, and 60 mg/kg in male quail produced significant hemolytic reactions with lymphocytosis, monocytopenia, and eosinophilia at 24 and 72 h after administration. Therefore, the necessary controls should be carried out for impurities of phenylhydrazine in certain drugs. The total amount of phenylhydrazine in human trials or dosing
If the maximum human dose is 2 g, the limit value for this impurity in the product is 0.05 mg/kg x 60 kg ÷ 2 g x 100% = 0.15% based on a human body weight of 60 kg.
(3) Extrapolation from safety studies For impurities for which safety data are unavailable, safety tests need to be carried out and then extrapolated from the NOAEL (no adverse effect level) data for the impurity.
In general, the following methods can be used.
①For example, the limit for impurity A in a preclinical trial sample of an innovative drug is 0.34%, and the maximum daily dosage is proposed to be 10 mg. The reporting entity has set the limit for impurity A at 0.5% for clinical study use. This limit exceeded the conventional impurity limit (0.1%) set by ICH Q3 and the limit of 0.34% for impurity A in preclinical samples. Therefore, in reviewing this variety, the Pharmacovigilance Centre recommended that the basis for determining this impurity limit be provided.
The investigator clarified this, and the derivation process was as follows: In the toxicity test in rats given repeatedly for 6 months, the NOA dose was 26.5 mg-kg-1-d-1, so the actual amount of impurity A was 26.5 x 0.34% = 0.09 mg-kg-1-d-1. This was converted by animal analogy to 0.09 mg-kg-1-d-1 x 6 = 0.54 mg-/m-2-d-1 ( a conversion factor of 6 for mg/kg dose in rats in humans) and then using the FDA conversion criteria ( a conversion factor of 37 for mg/kg vs. mg/m2 in humans), the amount of impurity A tolerated in humans equivalent to 60 kg is 0.54 x 60 ÷ 37 = 0.88 mg/d.
Considering that the maximum dose of this new drug in the clinical study was 10 mg if the impurity content of 0.5% is calculated, the maximum daily dose of impurity A is 0.05 mg/d, which is only 1/17 of the non-toxic dose of impurity A of 0.88 mg/d. Therefore, it is feasible to set the limit of impurity A at 0.5%.
②For example, the maximum daily dose of a drug administered orally is 60 mg, which corresponds to a dose of 1 mg/kg for a person weighing 60 kg. In a 13-week oral administration toxicology study in dogs, the NOAEL was determined to be 2 mg/kg.
Based on the 0.1% content of impurity A in this batch, the maximum safety limit for impurity A is 2 mg/kg x 0.1% = 2 μg/kg, which translates to a safe dose of impurity A in humans of approximately 1 μg/kg (conversion factor of 1.8 for mg/kg dose in dogs in humans), giving a limit of 1 μg/kg ÷ 1 mg/kg x 100% = 0.1%.
The above method is only a reference, and researchers can choose a reasonable method to develop scientific and reasonable impurity limits based on a combination of study results, which vary for different drug impurity limits.
Analytical methods
The choice of an analytical method is directly related to the specificity and accuracy of the impurity determination results. Therefore, the primary issue when conducting impurity studies is to choose the appropriate impurity analysis method.
1. Choice of analytical methods
(1) Analytical methods for organic impurities
The detection methods for organic impurities include chemical, spectroscopic, chromatographic, etc. Different detection methods are used depending on the structure of the drug and the degradation products. The impurities of different structures are separated and detected by suitable analytical techniques to control the impurities effectively. With the development and updating of separation and detection techniques, efficient and rapid separation techniques combined with sensitive, stable, accurate, and applicable detection methods, almost all organic impurities can be well separated and detected under suitable conditions.
In quality standards, the main impurity detection methods commonly used today are High-Performance Liquid Chromatography (HPLC), Thin Layer Chromatography (TLC), Gas Chromatography (GC), and Capillary Electrophoresis (CE). Suitable detection methods should be determined according to the physicochemical properties, chemical structure, and impurity control requirements of the drug and impurities.

As various analytical methods have certain limitations, attention should be paid to the mutual supplementation and validation between analytical methods of different principles, such as the mutual supplementation of HPLC and TLC and HPLC and CE, the mutual supplementation of reversed-phase HPLC systems and normal-phase HPLC systems, and the mutual supplementation of the detection results of different detectors of HPLC, etc. when carrying out impurity analysis.
(2) Analytical methods for inorganic impurities
The generation of inorganic impurities is mainly related to the production process. As many inorganic impurities directly affect the stability of the drug and can reflect the situation of the production process itself, it is important to understand the situation of inorganic impurities in the drug to evaluate the status of the drug production process. National pharmacopeias contain classical, simple, and effective detection methods for inorganic impurities.
For generic versions of mature manufacturing processes, the methods included in the pharmacopeia can be used to investigate and control the quality according to the actual situation. For new drugs produced by new production processes, the use of analytical techniques such as ion chromatography and inductively coupled plasma emission spectrometry-mass spectrometry (ICP-MS) is encouraged to carry out qualitative and quantitative analysis of various inorganic impurities that may be present in the product, to make a reasonable evaluation of its production process and provide a basis for the development of reasonable quality standards.
In general, non-volatile inorganic impurities are detected by the incineration residue method. Certain metal cationic impurities (silver, lead, mercury, copper, cadmium, bismuth, antimony, tin, arsenic, zinc, cobalt and nickel, etc.) are generally controlled by the heavy metal limit check method.
As lead is more frequently encountered in the production of pharmaceuticals. As lead is prone to accumulation and poisoning, it is used as a representative of heavy metals, and the limits for lead indicate heavy metal limits. Suppose limits are required for a specific metal ion(s) or metal ions that the above methods cannot detect. In that case, the more specific atomic absorption spectrophotometric method or the classical colorimetric method with a certain degree of specificity can be used (e.g., the detection of trace iron salts, ammonium salts, selenium, and other impurities in pharmaceuticals by the method of examination of iron salts, ammonium salts, and selenium already included in the pharmacopeia).
Although heavy metal detection methods can also detect arsenic, because of its toxicity and ease of bringing it into the product, it is necessary to use a highly sensitive and specific arsenic salt detection method for special investigation and control. The methods contained in various countries’ pharmacopeia have been effectively validated over the years and should be cited.
As sulfate ions, chloride ions, sulfur ions, etc., mostly come from the production of desiccants, catalysts or pH adjusters, etc., the investigation of their residues in the product can reflect the purity of the product, so the classical method in the pharmacopeia should be used for testing. If highly toxic substances (e.g., cyanide) are used in the production, the pharmacopeia method must detect trace residues that may be introduced into the product.
For the pharmacopeia has not been included in the detection of inorganic impurities (such as phosphate, phosphite, aluminum ions, chromium ions, etc.), according to its physical and chemical properties, the use of a certain degree of specificity, sensitivity, such as ion chromatography, atomic absorption spectrophotometry, colorimetric methods.
2, the validation of analytical methods
The validation of impurity detection methods should be carried out concerning the relevant technical guidelines, focusing on the validation of specificity and sensitivity. Specificity means that in the case of other components that may co-exist, the method used to determine the impurity characteristics is being measured accurately. The limit of detection is an important indicator of the sensitivity of the analytical method, and the limit of detection of the analytical method used must be by the requirements of the impurity limits in the quality standard, with the minimum limit of detection not being greater than the reported limit of the impurity.
To verify the specificity of the impurity analysis method, for API, according to its synthesis process, the intermediates of each step of the reaction (especially the last steps of the reaction intermediates), stereoisomers, crude products, recrystallization of the mother liquor, etc. as test products for systematic applicability study, to examine the product of the impurity peaks and the separation between the peaks of the main components are in line with the requirements, to verify the ability of the method to process the separation of impurities.
To investigate whether the method can effectively detect degradation products in APIs or formulations, suitable acid, base, light, heat, and oxidation reactions and other accelerated destructive tests can also be used to verify the specificity of the analytical method according to the chemical structure characteristics of the drug, the prescription and process of the formulation, storage conditions, etc. Diode array and mass spectrometry detectors can detect the purity of the peaks when necessary.
Because the degradation products produced under the forced degradation test conditions are more complex than those produced during the shelf life of the drug, with more unknown impurities and greater difficulty in separation, the above analytical methods can effectively show the purity of each chromatographic peak to avoid inaccurate analytical results as a result of the separation not meeting the requirements.
Suppose the test conditions for detecting the purity of the peaks are not available. In that case, the relative retention time of the peaks can be changed by appropriately adjusting the composition or ratio of the mobile phase, using the same test solution for accelerated destruction, and then comparing the number of impurity peaks before and after the adjustment of the mobile phase; the TLC method can also be used to compare the number and position of the spots of the same test solution for accelerated destruction under different unfolding systems. This is to demonstrate the specificity of the impurity analysis method.
The forced degradation test is essential for the separation of unknown impurities. It is designed to provide important information on separating impurities (especially degradates) from the main components, sample stability, and degradation pathways. During the test, care should be taken that destructive tests are moderate and should focus on examining acute conditions.
If the product is stable under certain conditions, there is no need to repeat the test by increasing the severity of the conditions. There is no uniform requirement for the degree of destructive testing at the moment, and it is generally appropriate that the content of the main components still accounts for the vast majority after strong destruction. At this point, a certain amount of degradation products have been produced, similar to the degradation of a sample left for a long time. It is more relevant to examine the degree of separation in this case.
Achieve this level of destruction requires mapping during the study, first through preliminary tests to understand the basic stability of the sample to light, heat, humidity, acid, alkali, and oxidation conditions, and then further adjustments to the destructive test conditions (e.g., light intensity, acid, and alkali concentration, time of destruction, temperature, etc.) to obtain results and profiles that adequately reflect the separation of the degradation products from the principal components.
In addition, comparing the change in the area of the main peak before and after the test, the relative response factor of the degradation products to the main component can also be roughly estimated. The stability of the sample under various conditions can be understood to provide information for selecting packaging and storage conditions, etc. If there is sufficient literature or test data for relatively stable drugs, then the forced degradation test can be waived.
3, Quantitative methods of organic impurities
The detection of organic impurities generally uses the HPLC method but sometimes also uses TLC, GC, and other methods.
The peak area method must be used if the HPLC method is used. The specific quantitative methods are
①External standard method (impurity control method)
② The main component control method with a correction factor, (iii) The main component control method without correction factor.
③ main component self-control method without correction factor.
④Peak area normalization method.
The ① method is a more accurate quantification method and should be used to assess and confirm controls and establish quality requirements.
The ② method requires a rigorous determination of the correction factor and is only suitable for controlling known impurities.
The ③ method assumes that the impurities have essentially the same response factors as the principal components. The response factors will not differ much if the impurity and the main component have similar molecular structures.
The ④ method is simple and quick, but there are limitations to its use in quality standards because the response factors of each impurity and principal component are not necessarily the same, the number of impurities and the number of principal components are not necessarily in the same linear range, and the instruments do not have the same integration accuracy and precision for trace impurities and constant principal components.
The substances in question include known impurities and unknown impurities. When the relative response factor of the known impurities to the principal components is within the range of 0.9-1.1 [3], the content can be calculated by the principal component’s control method; when it is beyond the range of 0.9-1.1, it is appropriate to calculate the content by the impurity control method, or by the principal component’s control method with correction factors. The ideal quantification method is a combination of the known impurity control method and the unknown impurity control method without correction factors. The researcher can choose the appropriate quantification method for the situation.
Technical factors such as production capacity and the feasibility of quality control should also be considered when selecting a suitable analytical method. Although the limits specified in the Annexes are accurate to the second decimal place, this does not mean that the analytical methods used in routine production quality control have to be so precise. Analytical methods such as thin-layer chromatography may also be used if the necessary validation has been carried out. If the analytical method is changed during the development process, a comparability study of the analytical results obtained before and after the method change should be carried out.
For TLC methods, the impurity control method and the main component’s control method are usually used for the control, the latter being used only if the color of the impurity spot is the same as the color of the main component spot.

Post time: Feb-09-2023



